Cleared in Months. Litigated for Years.
Regulatory strategy for surgical devices — where mesh and staplers taught the FDA that a fast clearance is not a safe one.
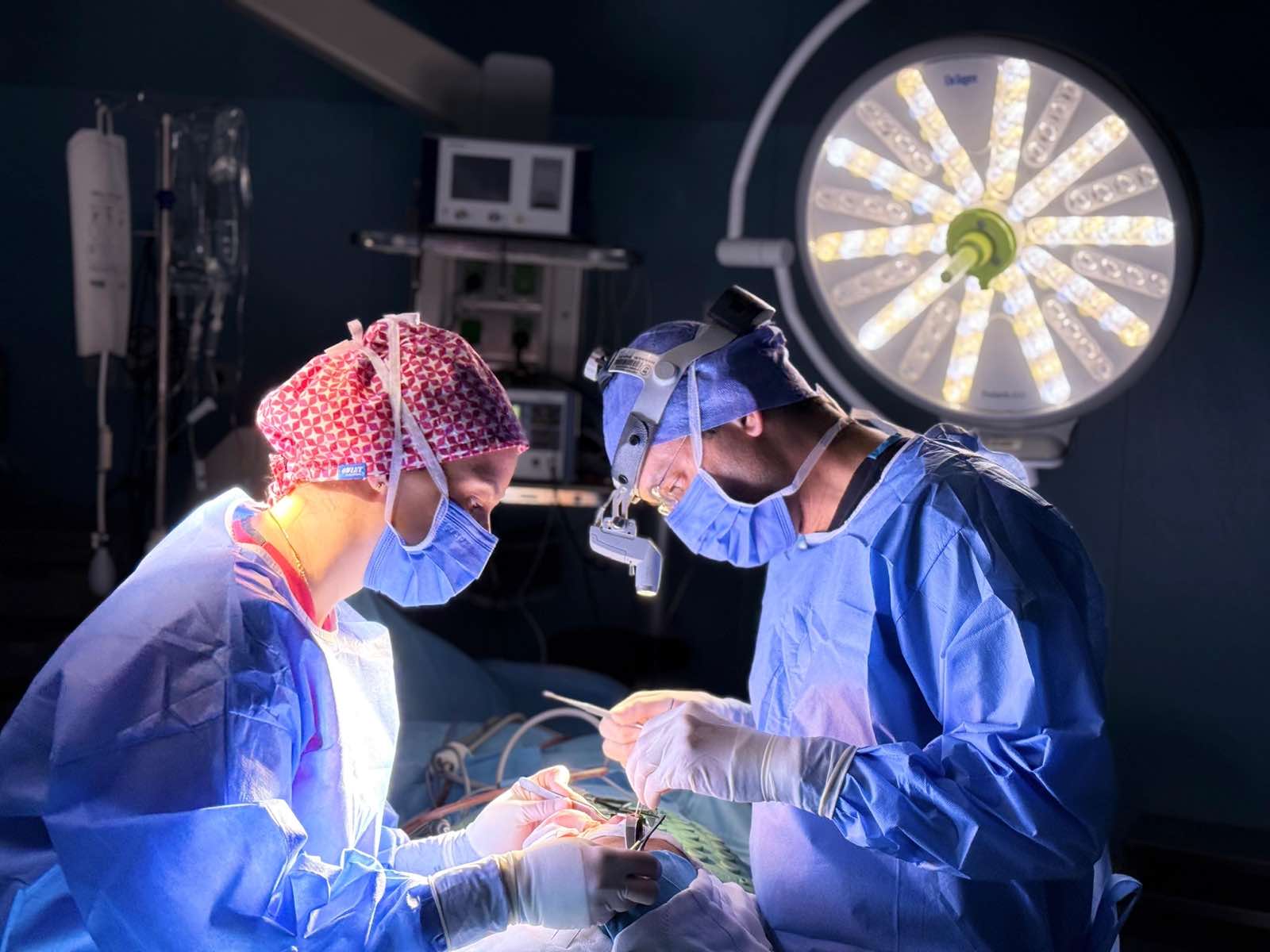
Surgical Devices Reach Patients Faster Than Almost Anything Else in Medicine.
Instruments, staplers, energy devices, and mesh mostly clear through 510(k) on substantial equivalence to a predicate — bench testing, biocompatibility, sterilisation validation, and no clinical trial. It is the fastest legitimate route to a patient's body that American regulation offers. That speed is the bargain, and the bargain has been tested.
Hernia mesh recalls, transvaginal mesh reclassification, and years of stapler malfunctions reported into a database almost nobody could see — each one taught the agency that predicate lineage does not guarantee safety. We build surgical programs that earn the fast route and survive what comes after it, because in this field the post-market is where the reckoning happens.
Bench data and a predicate put a device in a patient in months. The registry and the docket take years to answer.
Three Episodes That Rewrote Surgical Device Regulation.
Modern surgical regulatory expectations were not written in guidance first. They were written in recalls, reclassifications, and a reporting scandal — and every reviewer remembers them.
Hernia mesh
Predicate lineage is not safetyMeshes cleared on equivalence to predecessors that were themselves cleared on equivalence — until recalls and litigation exposed how far a device family can drift from the original evidence.
Transvaginal mesh
Reclassification is realThe FDA moved the devices to Class III, required PMAs, and ultimately ordered products off the market — proving the agency will revisit a class when the post-market data demands it.
Surgical staplers
Reporting cannot be hiddenMalfunctions were routed into a summary reporting programme largely invisible to the public. When it surfaced, the FDA ended the exemption and proposed reclassification.
The through-line is post-market evidence. All three episodes began with a clean premarket file and ended with a regulatory reversal driven by what happened afterwards. That is why a modern surgical submission is judged partly on the post-market surveillance and MDR discipline you bring with it — the reviewer is asking whether you will be the next case study.
Six Categories, One Reviewer With a Long Memory.
Instruments, implants, energy, and single-use economics each carry their own evidence bar within the same specialty.
Surgical Instruments
The deepest predicate space in devices, where clearance is fast and the real work is standards conformity and sterilisation validation.
Mesh & Implants
Where the field's hardest lessons live — long-term follow-up, material characterisation, and a reviewer who has seen this go wrong.
Closure & Stapling
Post-scandal territory: full MDR reporting, proposed reclassification, and human-factors expectations that keep rising.
Energy Devices
Thermal spread, tissue effect, and the surgical smoke question — bench-heavy submissions with real human-factors content.
Reusable Devices
Cleaning and sterilisation validation for complex geometries — the discipline duodenoscopes made a public-health issue.
Adjuncts & Biologics
Where a surgical adjunct crosses into biologic or combination-product territory and the lead center changes.
Under time pressure, in gloves, in blood. A misuse you did not test is a hazard you chose to accept.
A Device the Surgeon Misuses Is a Device That Failed.
The FDA's human factors expectations turned usability from a design nicety into submission content: use-related risk analysis, formative studies, and a validation study with representative users performing critical tasks. For surgical devices — used under time pressure, in gloves, in blood, by people who did not read the manual — this is not theatre.
The stapler experience made the point permanently. Many reported malfunctions involved use error as much as mechanical failure, and the agency's response treated the distinction as unhelpful. We build the human factors file as an evidence asset, because a foreseeable misuse you did not test is a hazard you accepted.
The Fast Route Is Real. So Is the Bill.
Surgical devices get to market quickly and are then judged for a decade by data the sponsor does not control.
is what a well-built 510(k) takes in a mature surgical predicate space — the fastest legitimate route to a patient in American regulation.
Transvaginal mesh moved to Class III and off the market; staplers lost their reporting exemption. The class is not permanent.
MDR discipline, complaint handling, and PMCF are now read as part of your premarket credibility, not as a separate obligation.
Six Failure Modes We Are Brought In to Prevent.
Speed is available in this field. It is also how most of the damage gets done.
Predicate drift
Building on a device that was itself cleared on equivalence to something else, until the lineage no longer supports the design in front of the reviewer.
Human factors as theatre
A validation study with the wrong users and no critical-task analysis, in a field where use error is the dominant failure mode.
Reprocessing underestimated
Complex reusable geometry with cleaning validation that will not survive post-duodenoscope scrutiny.
MDR discipline missing
Complaint handling and reporting that cannot demonstrate what the stapler episode made mandatory — visible, individual, timely reports.
Combination product surprise
A haemostat or sealant filed as a device when its biologic component makes it a combination product with a different lead center.
EU MDR clinical gap
Assuming FDA clearance travels, and meeting a notified body that wants clinical evaluation data the 510(k) never required.
Surgical Regulatory Leadership With Post-Market Memory.
Our surgical leads have run predicate strategy for instrument and implant families, built human factors files that held, and managed devices through reclassification.
"Surgery is the fastest route to market in devices and the least forgiving afterwards. Design the post-market evidence into the submission and the speed is yours to keep."
The discipline we bring across instruments, mesh, closure, energy devices, and reusable systems.
Bringing a Surgical Device Forward? Plan the Decade, Not the Clearance.
Bring senior surgical regulatory leadership in while the predicate, the human factors plan, and the post-market strategy are still one design.
Senior-led. Embedded in your team. No junior hand-offs.